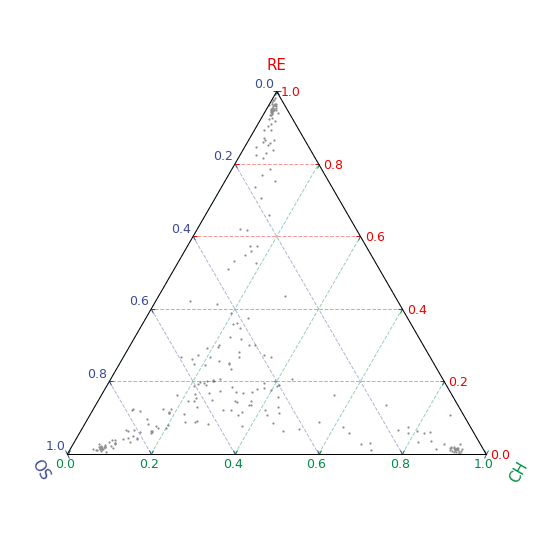
CytoSimplex.plot_ternary#
- CytoSimplex.plot_ternary(x, cluster_var, vertices, features=None, velo_graph=None, save_fig=False, fig_path='plot_ternary.png', fig_size=None, processed=False, method='euclidean', force=False, sigma=0.08, scale=True, title=None, split_cluster=False, cluster_title=True, dot_color='#8E8E8EFF', n_velogrid=10, radius=0.08, dot_size=0.6, vertex_colors=['#3B4992FF', '#EE0000FF', '#008B45FF'], vertex_label_size=12, gridline_alpha=0.4, axis_text_show=True, arrow_linewidth=0.004)[source]#
Create ternary plot that shows the similarity between each single cell and the three vertices of a simplex (equilateral triangle) which represents specified clusters. Velocity information can be added to the plot.
- Parameters
x (
Union[AnnData,ndarray,csr_matrix]) – Object that has the expression matrix of the single cells. Each row represents a single cell and each column represents a gene.cluster_var (
Union[str,list,Series]) – The cluster assignment of each single cell. If x is ananndata.AnnData, cluster_var can be a str that specifies the name of the cluster variable in x.obs. list orpandas.Seriesis accepted in all cases, and the length must equal to x.shape[0].vertices (
Union[list,dict]) –The terminal specifications. Should have exactly 3 elements for the 2-simplex (ternary simplex / triangle). Acceptable input include:
features (
Optional[list[str]]) – The features to be used in the calculation of similarity. It is recommended to derive this list withselect_top_features(). Default uses all features.velo_graph (
Union[csr_matrix,str,None]) – The velocity graph of the single cells, presented as an n-by-n sparse matrix. When x isanndata.AnnData, velo_graph can be astrto be used as a key to retrieve the appropiate data from x.uns. By default, no velocity information will be added to the plot.save_fig (
bool) – Whether to save the figure.fig_path (
str) – The path to save the figure.fig_size (
Optional[Tuple[int,int]]) – The size of the figure. The first number for width and the second for height.processed (
bool) – Whether the input matrix is already processed. When False, a raw count matrix (with integers) is recommended and preprocessing of library-size normalization, scaled log1p transformation will happen internally. If True, the input matrix will directly be used for computing the similarity.method (
str) – Choose from “euclidean”, “cosine”, “pearson” and “spearman”. When “euclidean” or “cosine”, the similarity is converted from the distance with a Gaussian kernel and the argument sigma would be applied. When “pearson” or “spearman”, the similarity is derived as the correlation is.force (
bool) – Whether to force the calculation when the number of features exceeds 500.sigma (
float) – The sigma parameter in the Gaussian kernel that converts the distance metrics into similarity.scale (
bool) – Whether to scale the similarity matrix by vertices.title (
Optional[str]) – The title of the plot. Not used when split_cluster=True.split_cluster (
bool) – Whether to split the plot by clusters. If False, all cells will be plotted in one plot. If True, the cells will be split by clusters and each cluster will be plotted in one subplot.cluster_title (
bool) – Whether to show the cluster name as the title of each subplot when split_cluster=True.dot_color (
str) – The color of the dots.n_velogrid (
int) – The number of square grids, along the bottom side of the triangle, to aggregate the velocity of cells falling into each grid.radius (
float) – Scaling factor of aggregated velocity to get the arrow length.dot_size (
float) – The size of the dots.vertex_colors (
list[str,str,str]) – The colors of the vertex labels, grid lines, axis labels and arrows.vertex_label_size (
int) – The size of the vertex labels.axis_text_show (
bool) – Whether to show the text along the axis.gridline_alpha (
float) – The alpha of the gridlines.arrow_linewidth (
float) – The linewidth of the arrows.
- Return type
Figure will be shown or saved.
Examples
import CytoSimplex as csx import scanpy as sc adata = sc.read( filename="test.h5ad", backup_url="https://figshare.com/ndownloader/files/41034857" ) vertices = {'OS': ["Osteoblast_1", "Osteoblast_2", "Osteoblast_3"], 'RE': ['Reticular_1', 'Reticular_2'], 'CH': ['Chondrocyte_1', 'Chondrocyte_2', 'Chondrocyte_3']} gene = csx.select_top_features(adata, "cluster", vertices) csx.plot_ternary(adata, "cluster", vertices, gene) csx.plot_ternary(adata, "cluster", vertices, gene, velo_graph='velo')